Reakcje rodnikowe – część 1/2
W chemii organicznej mamy do czynienia z jedynie 3 typami reakcji – polarnymi, pericyklicznymi oraz rodnikowymi. Wydawałoby się więc, że wystarczy poznać ogólne zasady rządzące każdym z tych typów i fajrancik. Na szczęście organicznych fanatyków, takich jak ja, wcale tak nie jest, więc mamy co robić. Najłatwiejszymi do opanowania (przynajmniej na poziomie olimpijskim) są reakcje rodnikowe i nimi się właśnie zajmiemy.
Zaczynając – reakcje rodnikowe to, jak sama nazwa wskazuje, takie, w których występują rodniki. A czym są te tajemnicze cząsteczki, znane powszechniej pod nazwą „wolne rodniki”? Definiujemy je jako cząsteczki zawierające niesparowane elektrony, a więc na pewno będą to cząsteczki o nieparzystej liczbie elektronów (jeśli posiadamy 2k+1 elektronów, w pary dobierzemy co najwyżej 2k z nich). Typowymi rodnikami, które spotykamy w nieorganice, są tlenek azotu(II) oraz (IV).
UWAGA – pod żadnym pozorem nie oznacza to, że cząsteczki o parzystej liczbie elektronów nie mogą być rodnikami – istnieje wiele rodników o dwóch niesparowanych elektronach, np. karbeny.
Rodniki mają to do siebie, że zazwyczaj nie lubią istnieć, więc chętnie zareagują z innymi cząsteczkami i przekażą im swoje problemy. Jest to dla nas nawet dobra wiadomość – możemy wygenerować w układzie małą ilość jakiegoś prostego rodnika, który zareaguje z większą cząsteczką, przekazując jej „rodnikowy charakter”. Związki, z których można w taki sposób wygenerować rodniki, nazywamy inicjatorami rodnikowymi. Poniżej dwa najczęściej stosowane inicjatory:
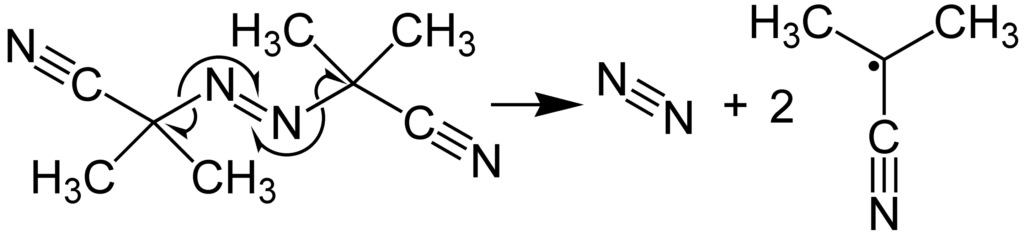
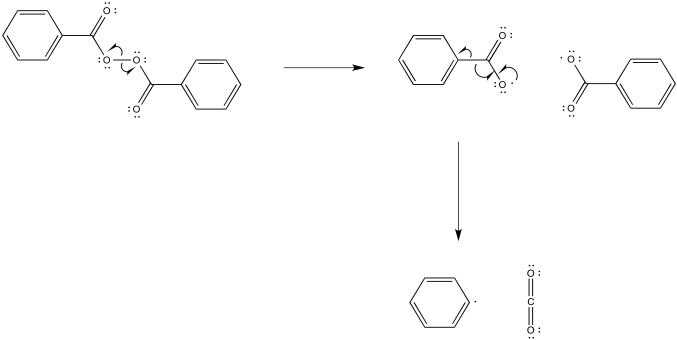
Innymi źródłami rodników są metaliczny lit/sód/potas w amoniaku bądź chlor/brom po naświetleniu:
Cl2 + hν → 2Cl•
Br2 + hν → 2Br•
Skoro mamy już metody generowania rodników, możemy zająć się reakcjami, jakie zachodzą pod ich wpływem. W pierwszej części tego artykułu omówię najważniejsze z punktu widzenia Olimpiady reakcje, a w drugiej reakcje mniej znane, ale moim zdaniem ciekawsze i równie użyteczne.
1. Chlorowanie/bromowanie w wersji klasycznej oraz allilowej/benzylowej
Klasyka gatunku – chlorowanie to w końcu jedna z niewielu reakcji, jakiej ulegają alkany. Ma ona jednak dwa ogromne problemy, przez które nie jest praktyczna. Primo, nie da się jej zatrzymać na jednokrotnej substytucji. Secundo, nawet jeśli jakieś warunki promują produkt monochlorowania, otrzymamy mieszaninę różnych produktów, o ile nie poddajemy reakcji wysoce symetrycznego związku:
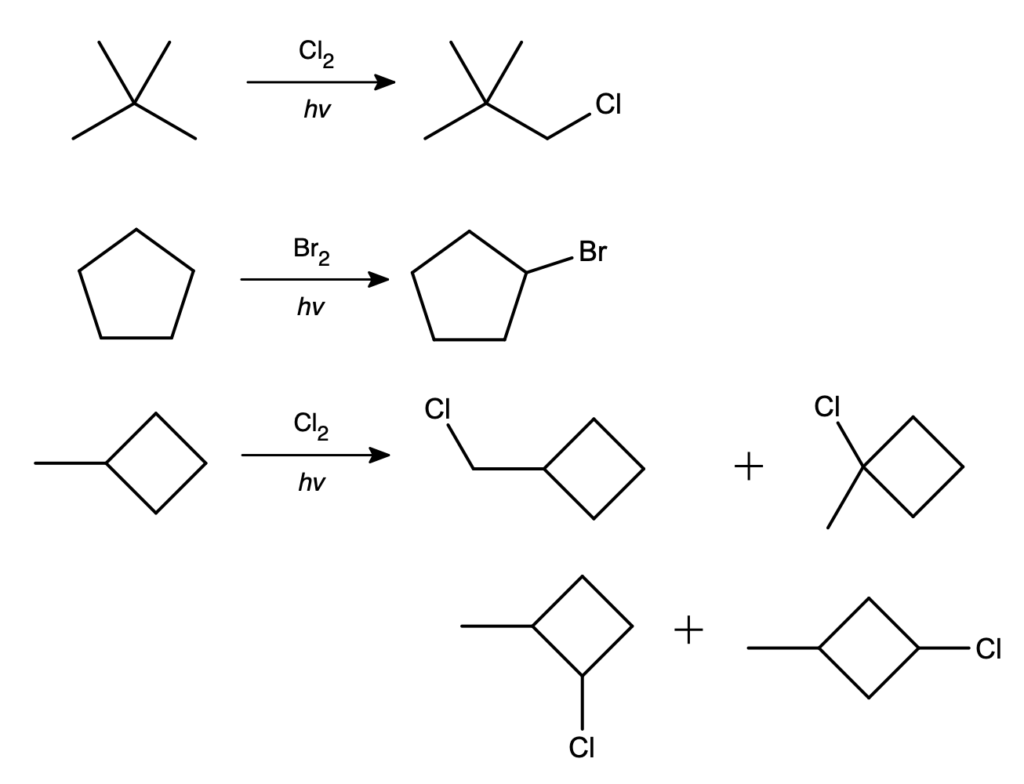
Co warto wiedzieć, reakcja ta wykazuje pewien stopień selektywności (bardziej widoczne w przypadku bromowania). Jeśli wgłębimy się w mechanizm reakcji, zobaczymy, że kluczowym i limitującym szybkość reakcji etapem jest oderwanie atomu wodoru i wytworzenie organicznego rodnika:
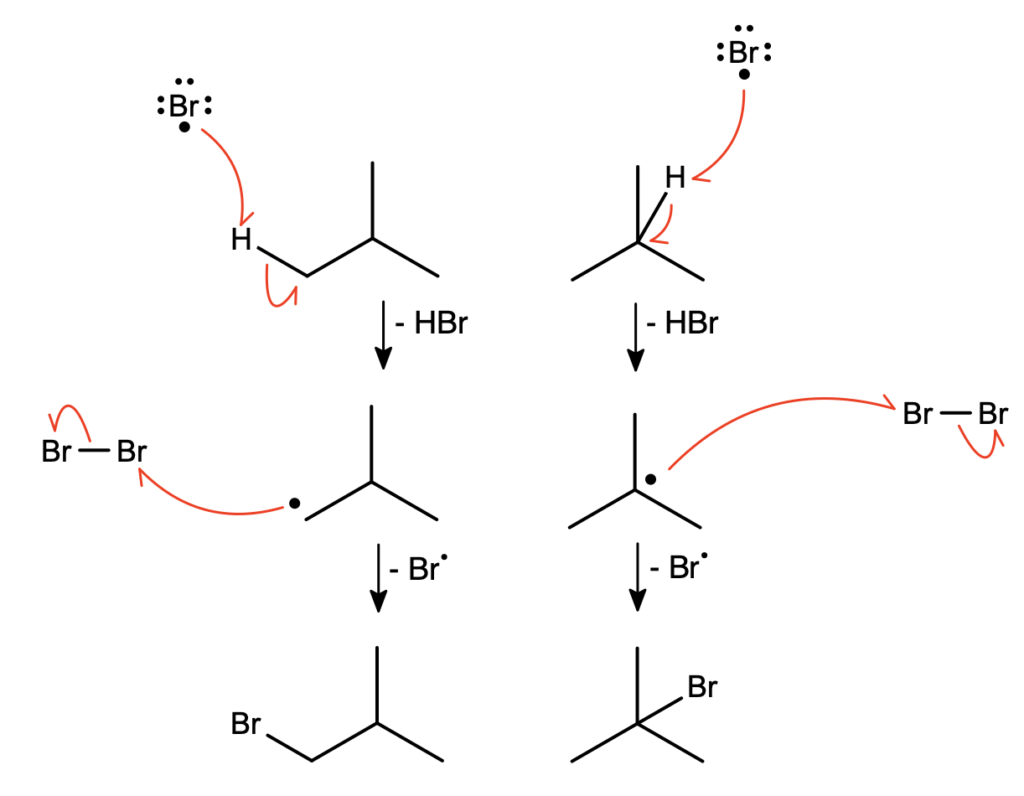
Najłatwiejsze do oderwania będą te atomy, które dadzą najbardziej stabilny produkt przejściowy. Jest to logiczne, ten produkt przejściowy, który mniej będzie chciał istnieć, trudniej będzie wytworzyć. A który z powyższych dwóch rodników będzie stabilniejszy? Zobaczmy, co pokazuje praktyka laboratoryjna:
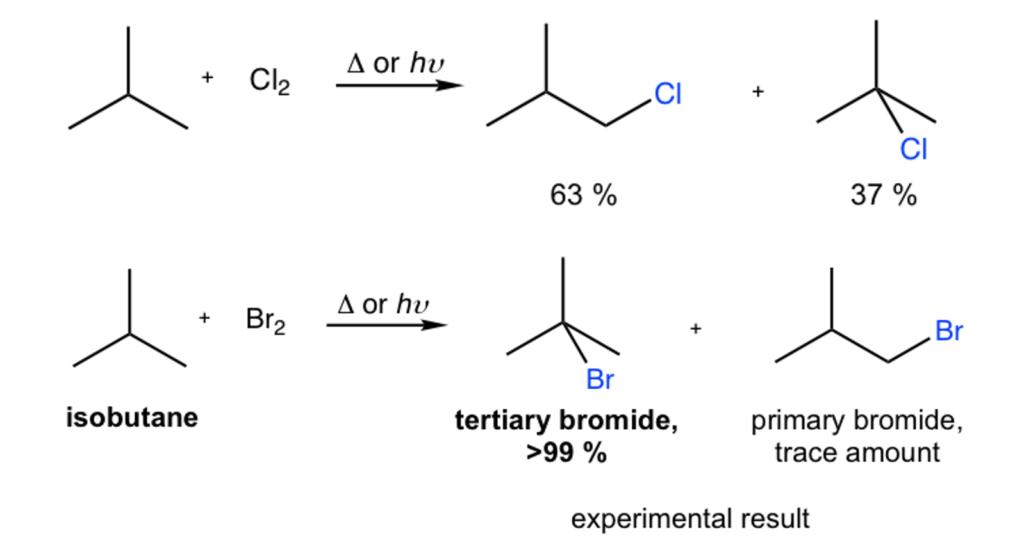
W przypadku reakcji bromowania doskonale widać, że o wiele stabilniejszy będzie rodnik trzeciorzędowy – produktu pierwszorzędowego powstają zaniedbywalne ilości. Przy chlorowaniu nie jest to już takie oczywiste, ale wystarczy sobie uświadomić, że pomimo 8-krotnego nadmiaru jednego typu atomów wodoru w izobutanie (1 wodór przy trzeciorzędowym węglu i 9 wodorów na trzech identycznych grupach -CH3), powstało jedynie 70% więcej produktu pierwszorzędowego. Selektywność jest więc o wiele mniejsza, lecz wciąż zauważalna.
Selektywność ta wynika ze zwiększonej stabilizacji rodnika w przypadku atomu węgla o wyższej rzędowości (tak zwana hiperkoniugacja – częściowe sprzężenie z sąsiednimi wiązaniami C-H; efekt indukcyjny grup alkilowych jest również istotny). W ten sposób zgrabnie przechodzimy do drugiego wariantu tej reakcji – bromowania allilowego/benzylowego. Ustaliliśmy przed chwilą, że im bardziej stabilizowany dany rodnik, tym większa selektywność. A co stabilizuje jeszcze lepiej niż wyższa rzędowość? Sprzężenie (bądź, jak kto woli, rezonans):
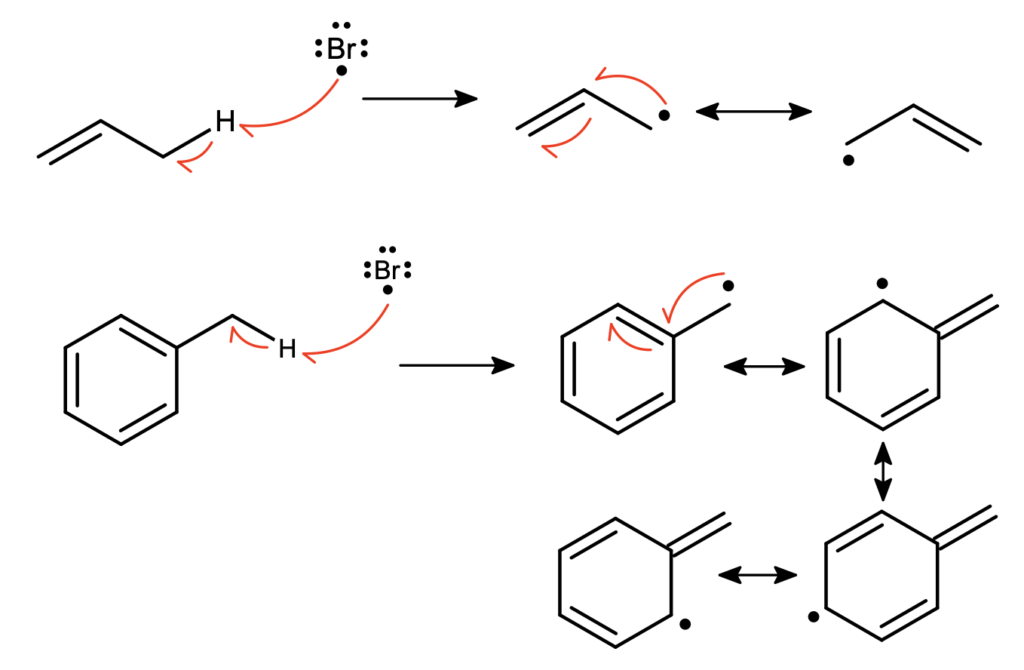
Jest to niezwykle użyteczna reakcja – pozwala bardzo selektywnie wprowadzić brom/chlor przy konkretnym atomie, z zadowalającymi wydajnościami. Przy bromowaniu allilowym jedynym problemem może okazać się konkurencyjna reakcja addycji bromu do wiązania podwójnego. Można go łatwo rozwiązać, używając zamiast bromu NBS (N-bromosukcynoimidu), pozwalającego na utrzymanie stałego, niskiego stężenia bromu. Istnieje również jego chlorowy analog – NCS.
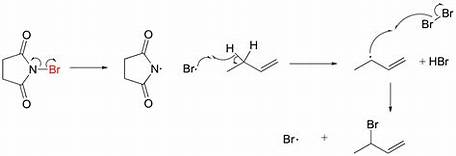
Kończąc o bromowaniu allilowym – należy cały czas pamiętać, że tu również może powstać kilka produktów – aby przewidzieć wszystkie możliwe z nich, narysuj każdą formę rezonansową. Najczęściej produktem dominującym będzie ten, w którym wiązanie podwójne będzie bardziej podstawione bądź sprzężone. Przy bromowaniu benzylowym nie ma tego problemu – pierścień nie ulega bromowaniu, gdyż straciłby swoją aromatyczność.
Spoiler alert!!! Nie patrz, jeśli nie robił*ś zadania 4. z tegorocznego pierwszego etapu. O wiele więcej nauczysz się, robiąc je samodzielnie.
W zadaniu należało – na podstawie szczątkowych informacji – zidentyfikować kilka izomerycznych węglowodorów. Jednym z nich był izobutylobenzen:
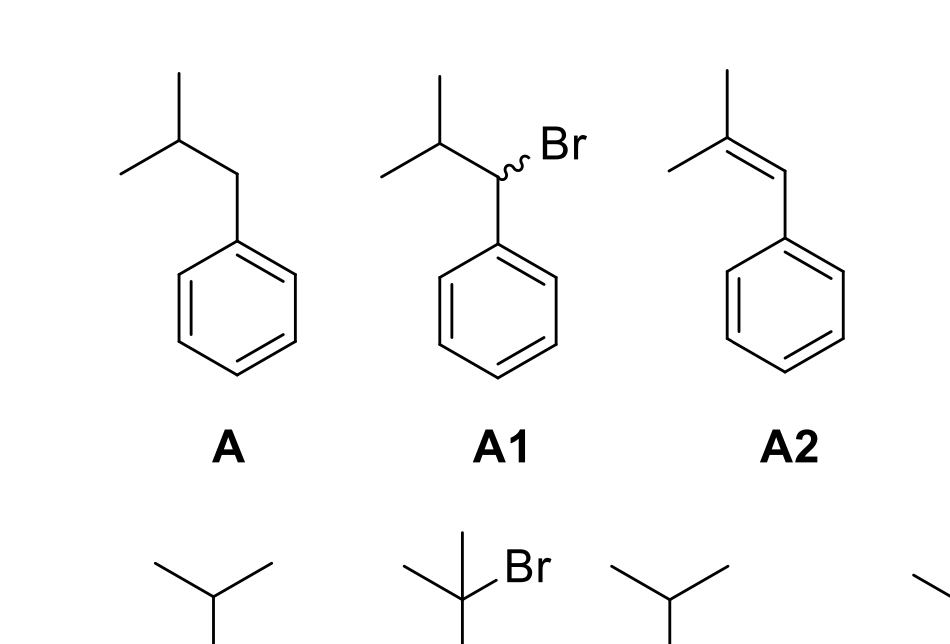
W tekście podana była informacja: „W tych warunkach z A powstaje jeden główny produkt monobromowania A1”. Autorowi chodziło o produkt monobromowania w pozycji stabilizowanej rezonansem, obok pierścienia benzenowego. Najprawdopodobniej umknął mu jednak fakt, że rodnik trzeciorzędowy jest równie stabilny i produkt bromowania w tej pozycji nie jest pomijalny, a nawet, jak pokazują dane doświadczalne, ilości obu produktów są zbliżone! Oczywiście, dało się wydedukować i potwierdzić poprawną strukturę na podstawie innych informacji, ale ta nieścisłość wprowadziła w błąd i pozbawiła punktów wielu zawodników.
2. Reakcje redukcji litowcami
Nie, nie będzie nic o memicznej reakcji Wurtza – zajmiemy się reakcjami mającymi jakiekolwiek znaczenie praktyczne. Porozmawiamy o tak zwanych „dissolving metal reductions” (po polsku „roztwarzający metal redukcje”, ale nie brzmi to najlepiej).
Znasz pewnie doskonale reakcje wypierania wodoru z kwasów przez metale:
Zn + 2HCl → ZnCl2 + H2
To samo dzieje się w słabych kwasach, jak kwas octowy, woda czy nawet… amoniak. Może on nie wyglądać Ci na świetny kwas, ale raczej na typową zasadę. Ale w rzeczywistości może on pełnić rolę zarówno kwasu, jak i zasady – jest związkiem amfiprotycznym. Sumaryczną reakcję wypierania wodoru z amoniaku możemy zapisać jako:
2Na + 2NH3 → 2NaNH2 + H2
Nie wyjaśnia to jednak istoty reakcji – nie obserwujemy natychmiastowego wydzielania się stechiometrycznej ilości wodoru, lecz błękitne zabarwienie roztworu:

Okazuje się, że przy niskich stężeniach metalu uwolnione elektrony wcale nie muszą od razu zredukować amoniaku. W roztworze takim istnieją solwatowane kationy Na(NH3)6+ (podobne do hydratu, ale z amoniakiem, więc to amoniakat) oraz analogicznie solwatowane elektrony e(NH3)6– znajdujące się w odpowiednio dużych lukach rozpuszczalnika. Związki takie nazywamy elektrydami. Dopiero po czasie elektrony redukują atomy wodoru z amoniaku. Zadanie z Olimpiady o elektrydach.
Jeśli jednak w roztworze jest obecny łatwiejszy do redukcji związek, możliwe jest okiełznanie elektronów i użycie ich do zredukowania go. Najpopularniejszą tego typu reakcją jest redukcja Bircha, od nazwiska Arthura Bircha. W odmętach internetu można jednak znaleźć nazwę „redukcja Brzozy” 
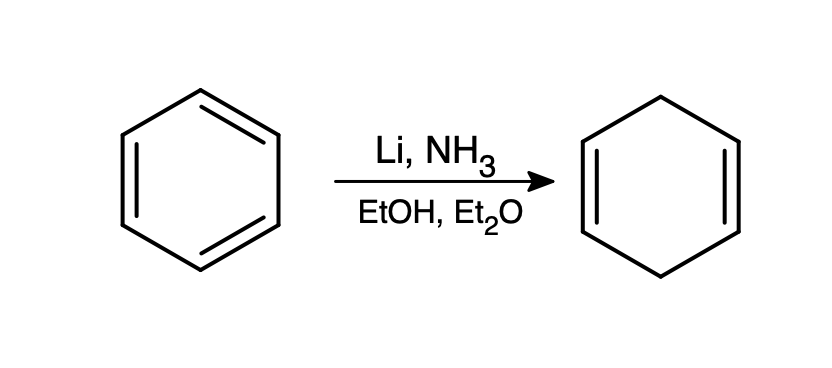
Pierścień aromatyczny jest lepszym akceptorem elektronów niż amoniak – antywiążący orbital LUMO jest względnie niskoenergetyczny przez delokalizację. Powstały w ten sposób anionorodnik jest mocno zasadowy i odrywa proton od etanolu. Zabawa z dostarczeniem elektronu, a następnie protonu, powtarza się jeszcze raz, w wyniku czego otrzymujemy niesprzężony cykloheksa-1,4-dien:
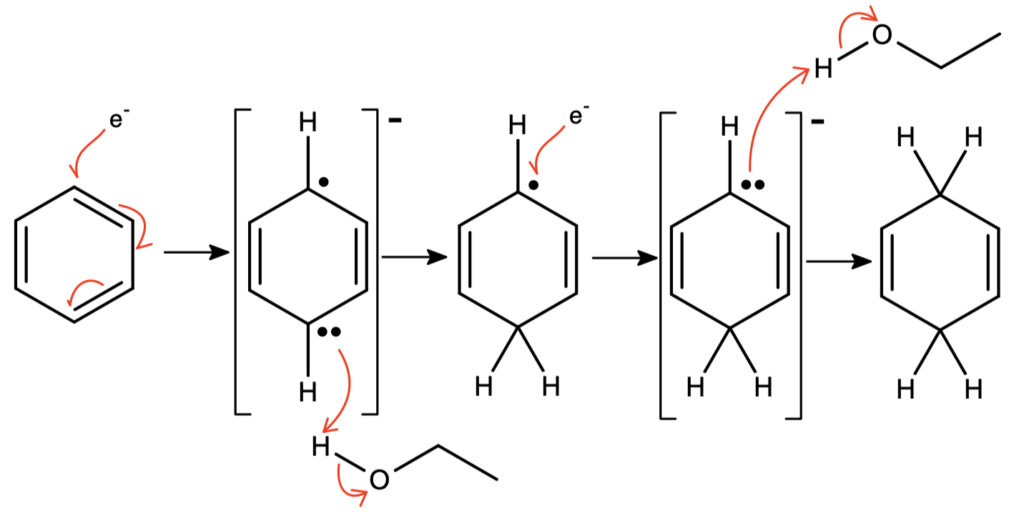
Uważniejszy czytelnik zada sobie pytanie – dlaczego nie powstaje trwalszy, bardziej sprzężony cykloheksa-1,3-dien, skoro anion w ostatnim etapie jest zdelokalizowany? Zadawałem sobie to samo pytanie – żaden podręcznik do chemii organicznej nie był w stanie na nie odpowiedzieć; odpowiedź wymaga wiedzy z poziomu wyższego, niż prezentowany w tych podręcznikach (a także w tym poście). Żądnych odpowiedzi odsyłam do publikacji omawiającej obliczenia kwantowe w chemii organicznej, strony 126-128 (analogie do anionu cyklopentadienowego).
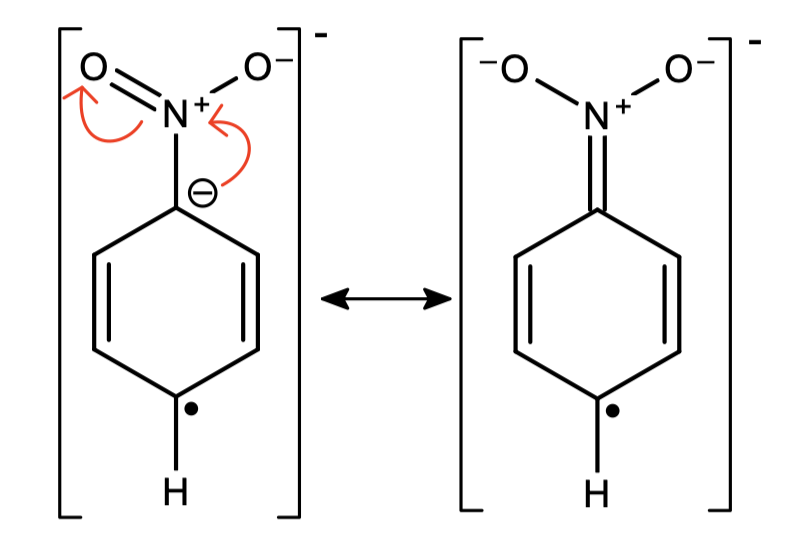
Redukowanie „gołego” benzenu nie jest zazwyczaj potrzebną reakcją – co, jeśli przyłączone są do niego jakieś podstawniki? W przypadku tylko jednopodstawionego pierścienia teoretycznie możliwe są dwa produkty: redukcji w pozycjach ipso-para lub orto-meta:
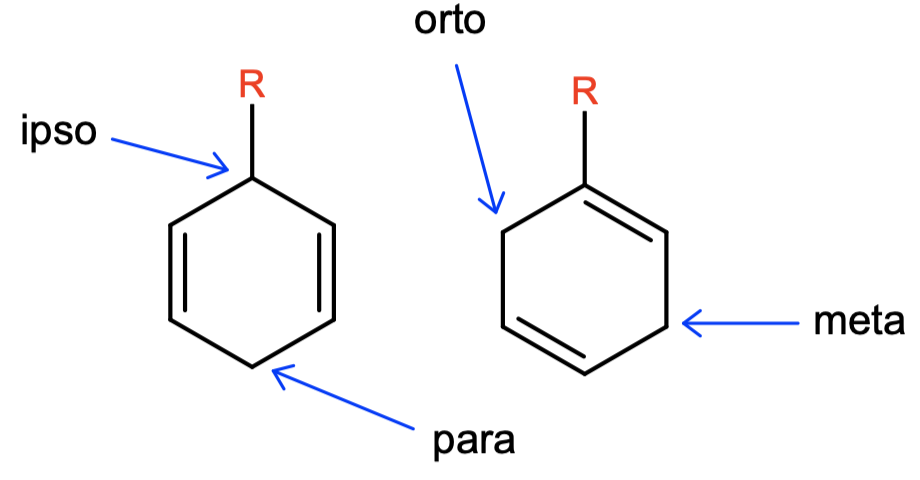
Okazuje się, że jeśli -R to grupa wyciągająca elektrony, np. -COOH, -NO2, to otrzymamy jedynie produkt ipso-para. Pierścienie z grupą donorującą elektrony, jak -OCH3 czy -N(CH3)2, zredukują się w pozycjach orto-meta. Dzieje się tak przez stabilizację ładunku ujemnego w odpowiednich miejscach (grupy wyciągające) bądź destabilizację (grupy donorujące).
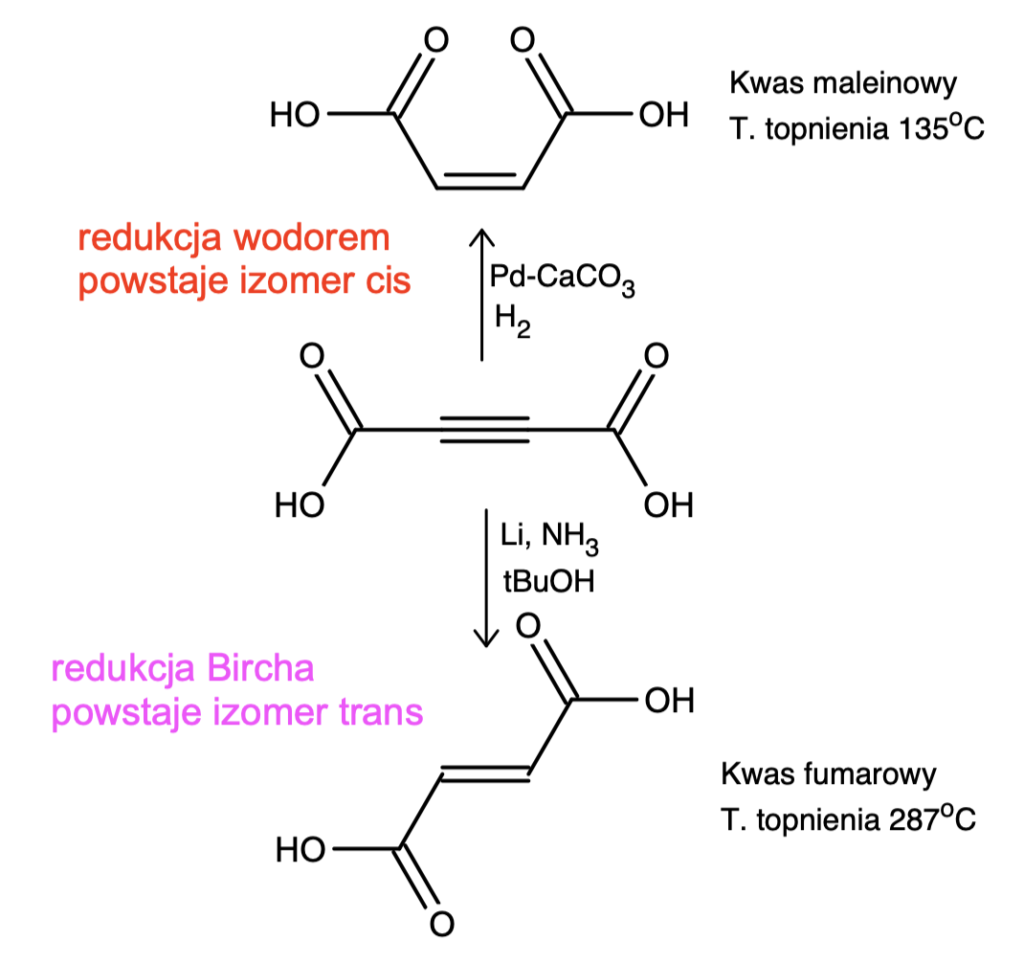
Redukcja Bircha jest tak użyteczną reakcją, ponieważ można ją wykorzystać nie tylko do redukcji pierścieni aromatycznych, ale też alkinów. Pozwala ona na ich selektywne zredukowanie do alkenu trans, co wraz z komplementarną reakcją redukcji wodorem na katalizatorze Lindlara daje możliwość otrzymania alkenu o dowolnej konfiguracji wiązania podwójnego z odpowiedniego alkinu:
Mechanizm tej reakcji przebiega całkiem podobnie do redukcji pierścienia, następują kolejno addycje elektronu oraz protonu:
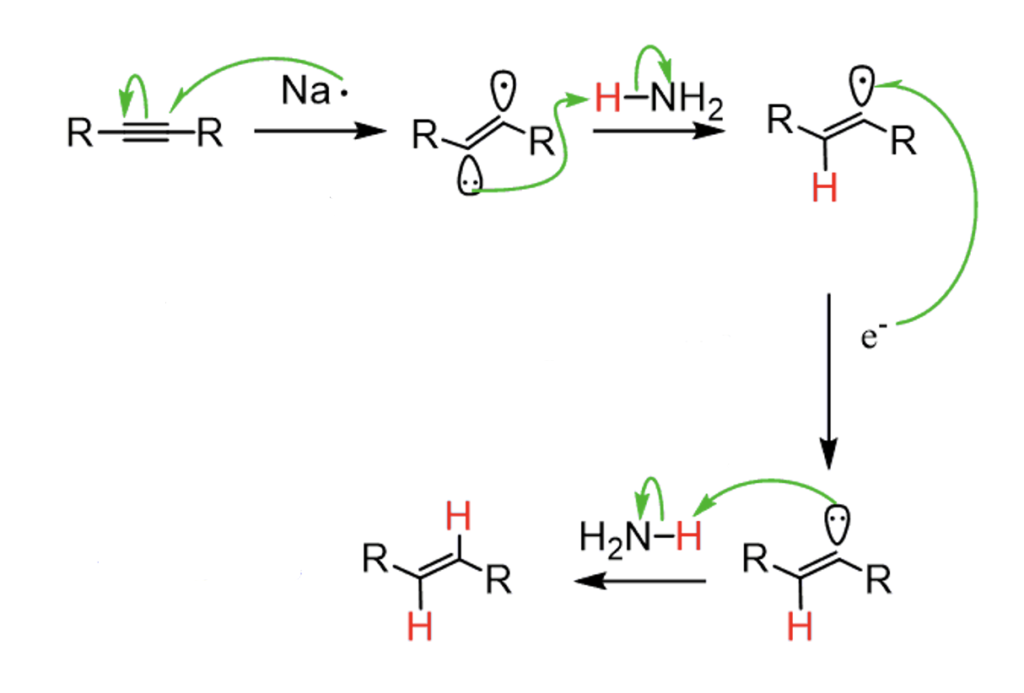
Najważniejszą cechą tej reakcji jest otrzymywanie produktu jedynie o konfiguracji trans. Nasuwa się ważne pytanie – z czego ona wynika? Odpowiedź jest o wiele bardziej złożona, niż mogłoby się wydawać. W wielu źródłach można znaleźć błędną informację, że przez obecność dodatkowych elektronów możliwy jest swobodny obrót wokół wiązania podwójnego w anionorodniku – dawałoby to rzeczywiście trwalszy produkt trans. Lecz obrót ten nie jest możliwy – to wciąż silne i niezależne wiązanie podwójne!
Prawdę ukazuje nam dopiero DFT – teoria funkcjonału gęstości, wykorzystująca metody kwantowe, pozwalające na m.in. porównywanie prawdopodobnych mechanizmów i wybieranie lepszego. Okazuje się, że różnica w energiach pomiędzy wiązaniem cis a trans jest dosyć spora, ale występuje pomiędzy nimi jeszcze jeden stan – cząsteczka liniowa. Dochodzi do rehybrydyzacji anionorodnika (sp zamiast wcześniejszego sp2) i na odwrót. Nie ma tu więc mowy o swobodnym obrocie wiązania, a raczej o jego „przełączaniu” pomiędzy trzema stanami. Bardziej wnikliwych czytelników odsyłam do dwóch publikacji w przypisach.
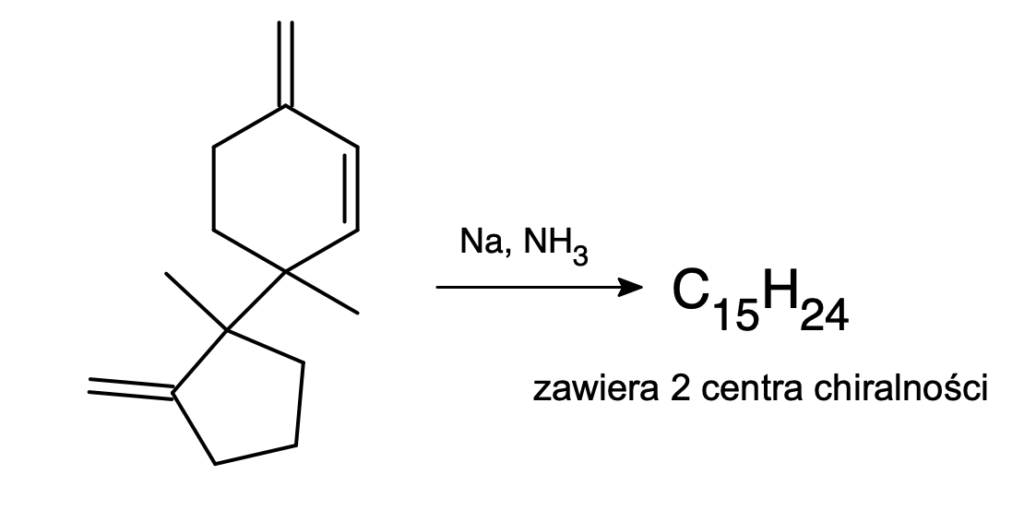
Na koniec średniej trudności zagadka o nieco nietypowym zastosowaniu redukcji Bircha – jest to ostatni etap syntezy ciekawego związku wyizolowanego z grzyba:
Źródła do dalszego poczytania:
- March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, dział 14
- Molecular Orbitals and Organic Chemical Reactions; strony 126-128
- Dissolving metal reduction of acetylenes: a computational study; DOI: 10.1021/jo061583j
- First observation of alkyne radical anions by electron spin resonance spectroscopy: Hexyne/n‐hexane mixed crystals; https://doi.org/10.1063/1.460659
Komentarze |0|
Tagi: Alkeny, Alkiny, Elektrydy, Halogenowanie, Mechanizm, Metaloorganika, Redukcja, Rodniki, Zadanie we wpisie, Związki aromatyczne