Reakcje rodnikowe – część 2/2
Wjeżdżamy w nowy rok szkolny długo wyczekiwaną kontynuacją naszego pierwszego wpisu. Link do części 1/2 znajdziesz tutaj.
Ciąg dalszy rodnikowych peregrynacji. Zgodnie z zapowiedzią, w tej części zobaczysz rzeczy mające mniejszą szansę pojawić się na OlChemie, ale ciekawsze i nie mniej istotne w praktyce. Tym razem postaram się je omówić inaczej – dotrzemy do konkretnych reakcji, wykonując proste eksperymenty myślowe na dobrze znanych Ci już konceptach. Właśnie tak zazwyczaj odkrywane są rewolucyjne reakcje – poprzez odkrywanie nowych powiązań między znanymi faktami, a następnie próby zrealizowania tego w praktyce, a nie przez mieszanie losowych odczynników.
1. Co jeszcze może „zwabić” wolne elektrony?
Poprzednio skończyliśmy na redukcjach i od tego też zaczniemy.
Rozważaliśmy reakcje, gdzie solwatowane elektrony pełnią funkcję nukleofila. Były to jednak dosyć nietypowe przykłady “addycji” nukleofilowej, gdyż do wiązań wielokrotnych węgiel-węgiel. Można więc przypuszczać, że jeśli w podobny sposób przereagowalibyśmy grupę „znaną” przede wszystkim z addycji nukleofilowych do niej, otrzymamy analogiczny produkt. Znasz takie grupy?
Od razu narzuca się karbonyl – wszelkie grupy karbonylowe są znane z podatności na addycję nukleofilową. Zacznijmy od najmniej złożonego przypadku, czyli ketonów i aldehydów. Po potraktowaniu ich metalicznym sodem oraz źródłem protonów (w tym przypadku metanolem), obserwujemy dokładnie to, czego się spodziewaliśmy! Reakcja ta nosi nazwę redukcji Bouveault–Blanc.
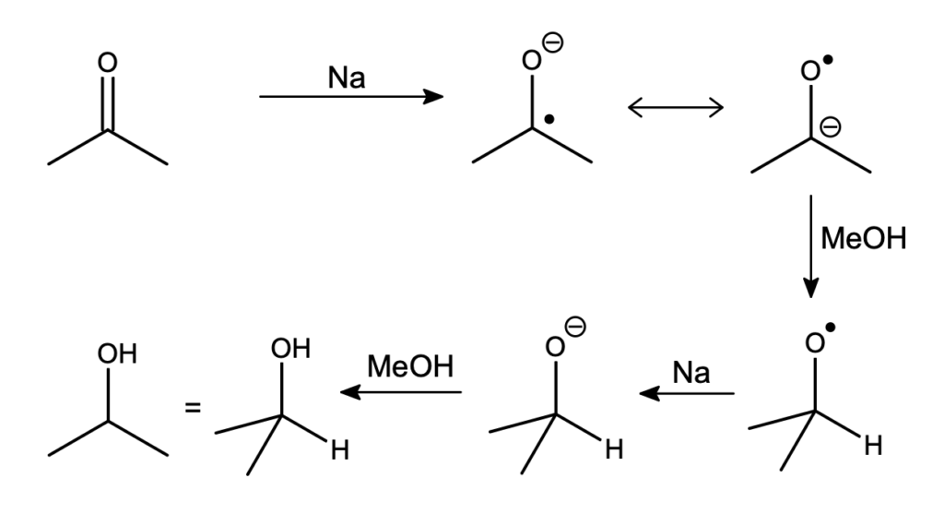
“Chwila, miały być fajne i użyteczne reakcje”. Być może nie jest to najfajniejsza reakcja – w końcu mało kto chciałby mieć do czynienia z metalicznym sodem (ryzyko zapłonu), a do analogicznej redukcji mamy lepsze, bezpieczniejsze i o wiele selektywniejsze odczynniki, jak np.  .
.
Prowadźmy jednak nasze rozumowanie dalej, wgłębiając się w mechanizm tej redukcji i myśląc, co można by w nim zmienić, aby osiągnąć coś nowego.
Pamiętajmy, że rodniki mogą przereagować w celu zmniejszenia swojej reaktywności na 3 sposoby:
- z cząsteczkami innych związków;
- wewnątrzcząsteczkowo (uwalniając mniejszą cząsteczkę lub się przegrupowując);
- z innymi cząsteczkami (tego samego) rodnika, np. dimeryzując.
Z ostatnią opcją mamy do czynienia najrzadziej – w środowisku nie mogą być obecne inne reaktywne cząsteczki, a stężenie rodnika musi być dosyć duże. Takie warunki nie są jednak niemożliwe – wystarczy przeprowadzić poprzednią reakcję, lecz nie dodawać (przynajmniej na początku) źródła protonów:
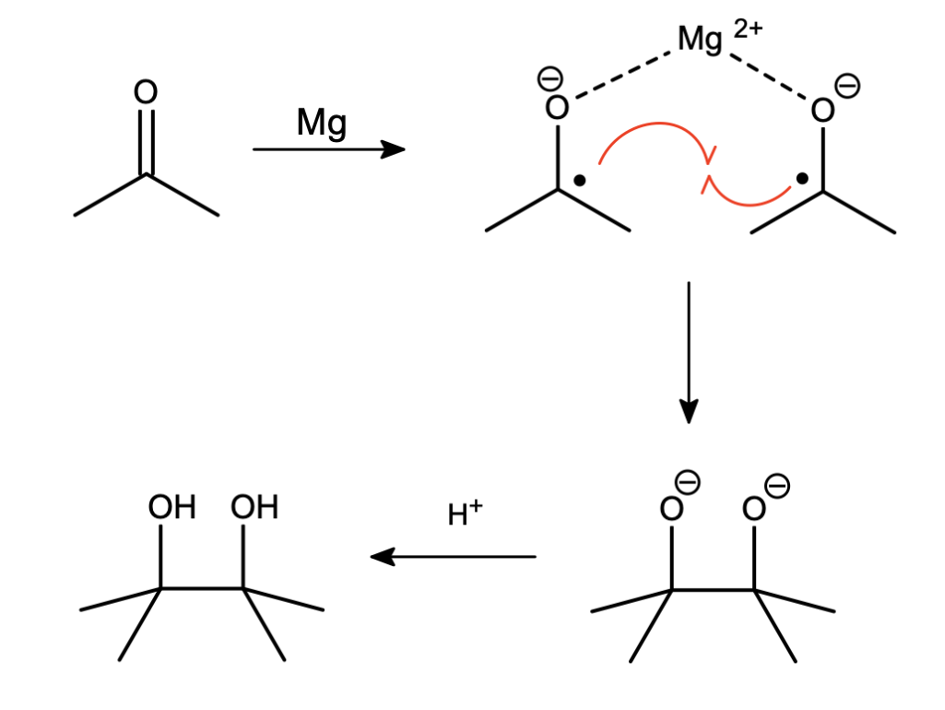
Analogiczna do wcześniejszej addycja elektronów daje anionorodnik acetonu, który przy nieobecności źródła protonu dimeryzuje. Na koniec work-up z użyciem np. metanolu bądź wody.
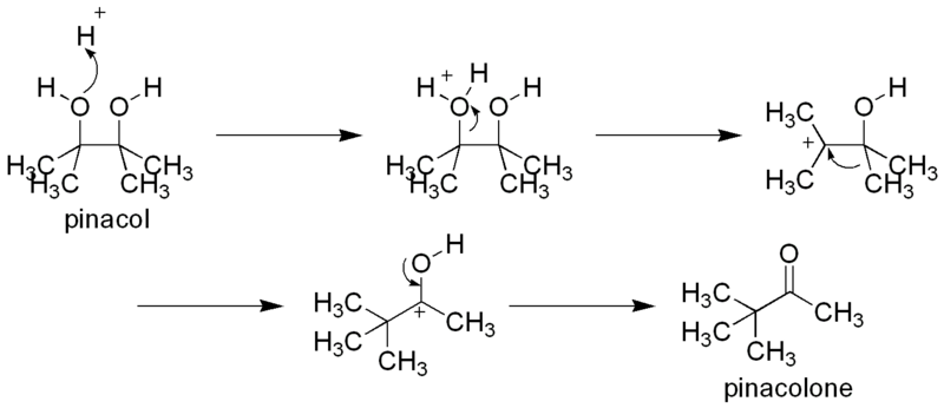
Jest to bardzo ciekawa i przydatna reakcja, nosząca nazwę sprzęgania pinakolowego – powyżej widzisz jej pierwszy zaobserwowany wariant. Nazwa reakcji pochodzi od produktu który powstaje, gdy jako substratu użyjemy acetonu – pinakolu. Być może znasz go z przegrupowania pinakolowego:
Jak się okazuje, estry mogą również ulegać podobnemu sprzęganiu, prowadzącemu do trochę innych produktów.
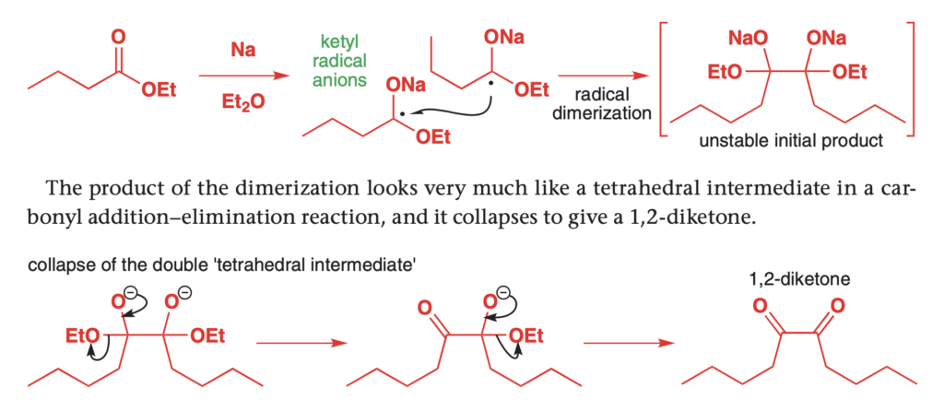
Reakcji niestety nie da się zatrzymać na tym etapie, 1,2-diketony są znacznie bardziej elektrofilowe od estrów. Do kolejnej redukcji elektronami dochodzi znacznie szybciej niż do sprzęgania, użycie stechiometrycznej ilości sodu więc nie pomoże. Jako produkt końcowy otrzymuje się hydroksyketony:
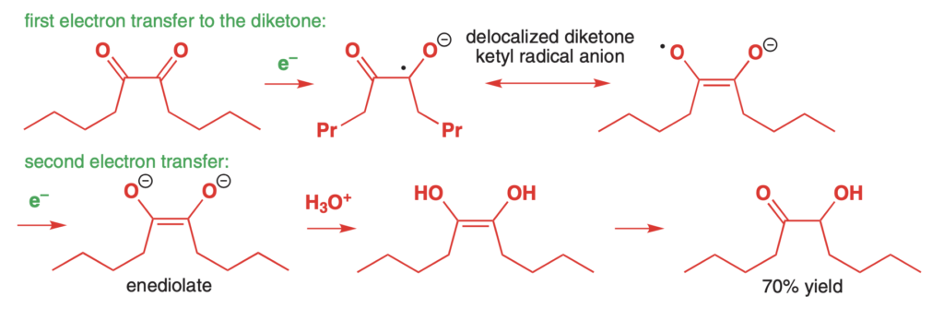
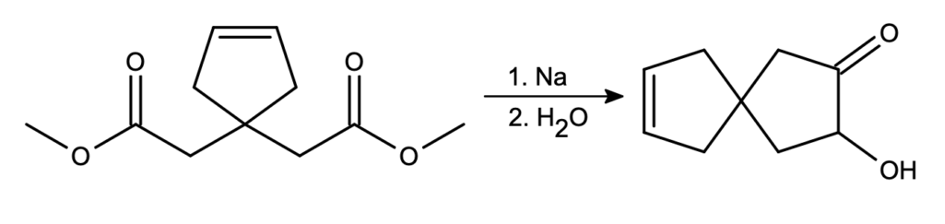
Reakcji tej można z powodzeniem używać do zamykania dowolnej wielkości pierścieni, jeśli w cząsteczce nie są obecne inne podatne na redukcję grupy funkcyjne:
Nowocześniejszą modyfikacją tego typu sprzęgań jest reakcja McMurry’ego (tak, tego McMurry’ego od popularnych podręczników). Jako reduktora używa się mieszaniny  albo
albo  oraz
oraz  . W ten sposób generowany in situ (generowanie substratu/katalizatora bezpośrednio w środowisku reakcji) jest metaliczny tytan. Trzeba mieć na uwadze jedną różnicę – w opisanej reakcji diolowego produktu nie da się wyizolować – powstaje odpowiedni alken. Można to próbować wyjaśnić dużym powinowactwem tlenu do tytanu, niemniej mechanizm przejścia z diolu w alken nie jest do końca znany, przemiana zachodzi na powierzchni metalu.
. W ten sposób generowany in situ (generowanie substratu/katalizatora bezpośrednio w środowisku reakcji) jest metaliczny tytan. Trzeba mieć na uwadze jedną różnicę – w opisanej reakcji diolowego produktu nie da się wyizolować – powstaje odpowiedni alken. Można to próbować wyjaśnić dużym powinowactwem tlenu do tytanu, niemniej mechanizm przejścia z diolu w alken nie jest do końca znany, przemiana zachodzi na powierzchni metalu.
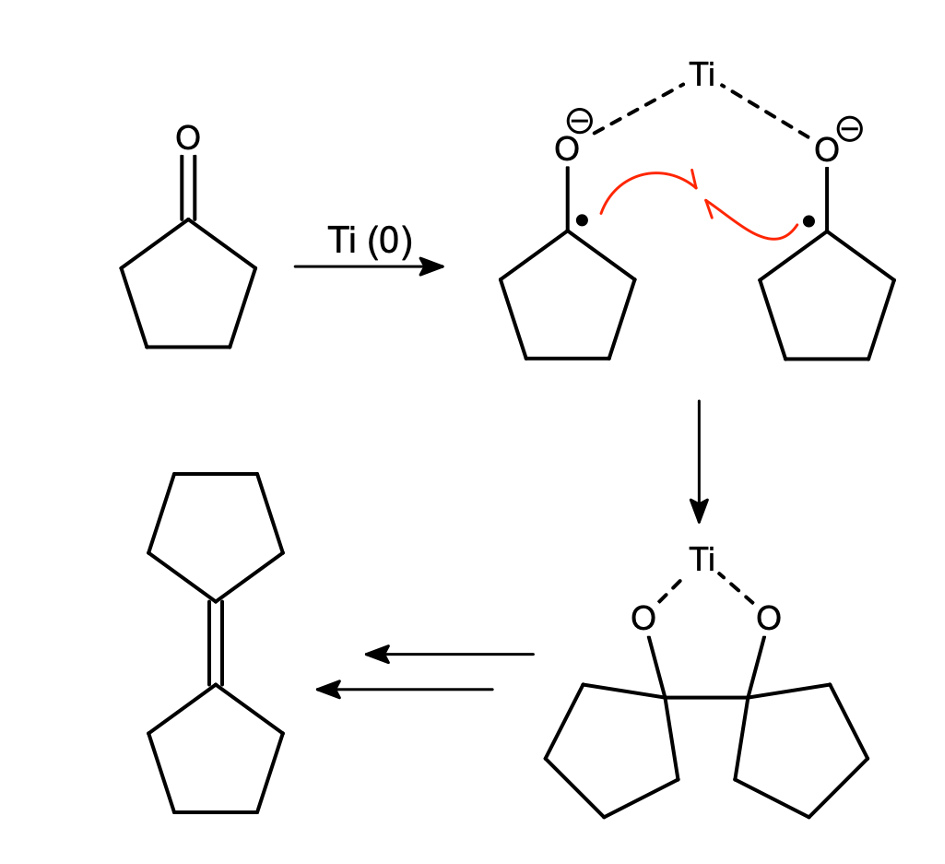
Reakcja ta wykazuje trochę większą tolerancję na inne grupy funkcyjne, przy użyciu modyfikowanych warunków możliwe jest również sprzęganie estrów, a nawet amidów. Międzycząsteczkowe, mieszane reakcje McMurry’ego (takie, w których reagują ze sobą dwa różne związki karbonylowe) zazwyczaj nie kończą się powodzeniem, gdyż dają mieszaninę produktów.
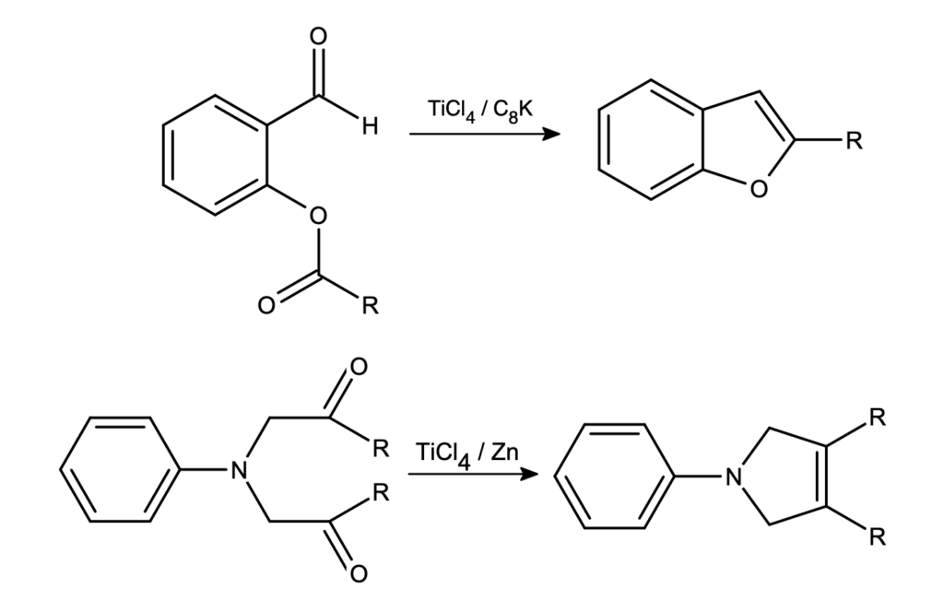
Reakcje tego typu należą jednak do rzadkości, rodników zazwyczaj jest w środowisku reakcji zbyt mało, by mogły ze sobą (w zauważalnym stopniu) reagować. Niezbyt obchodzi je z czym przereagują, czy będzie to rozpuszczalnik, czy inna, losowa cząsteczka. Znacznie częściej będą inicjować reakcje łańcuchowe – jednej z nich (bromowaniu) przyjrzeliśmy się ostatnio. Teraz czas na nowocześniejsze, bardziej selektywne i użyteczne reakcje tego typu.
2. Wstęp do chemii cynoorganicznej
Przy planowaniu syntezy często dochodzi do sytuacji, gdy musimy pozbyć się zbędnego atomu chlorowca – zazwyczaj ma to miejsce po addycji np. bromu i wykorzystaniu go do tego, co chcieliśmy osiągnąć:
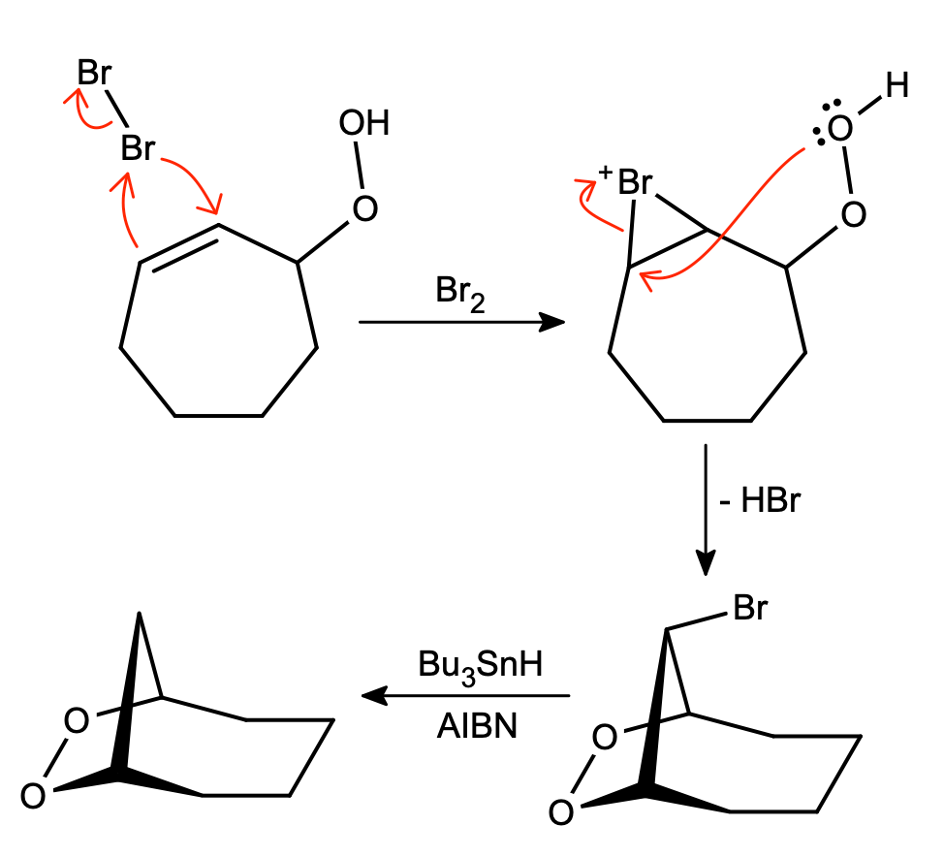

Jest to niezwykle selektywna reakcja – nie naruszyła nawet mostka tlenowego, który często odpowiada za właściwości wybuchowe organicznych nadtlenków. Możliwe jest także usunięcie atomu bromu, gdy w cząsteczce obecne są atomy chloru. Jak nietrudno się domyślić, skoro post jest o reakcjach rodnikowych, pierwszym etapem tej reakcji będzie wygenerowanie rodnika. Podczas ogrzewania mieszaniny zawierającej  powstaje azot oraz rodnik stabilizowany grupą nitrylową (patrz część 1/2).
powstaje azot oraz rodnik stabilizowany grupą nitrylową (patrz część 1/2).
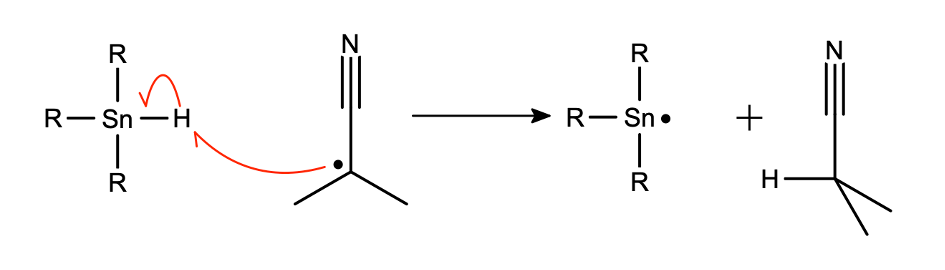
Rodnik odrywa atom wodoru, wytwarzając nowy rodnik –  :
:
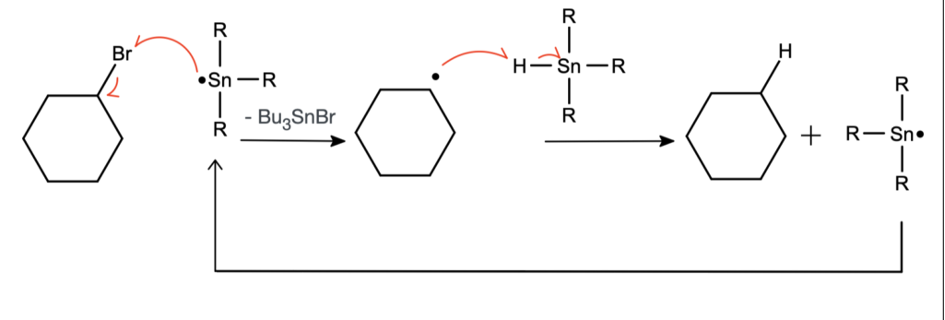
Następnie dochodzi do oderwania atomu bromu przez wygenerowany rodnik cynowy. Powstaje w ten sposób oraz rodnik organiczny, który odrywa atom wodoru od innej cząsteczki  . Wytwarza się w ten sposób kolejny rodnik i reakcja jest powtarzana.
. Wytwarza się w ten sposób kolejny rodnik i reakcja jest powtarzana.
 jest jednocześnie zużywane jako źródło atomów wodoru oraz propagator rodników, dlatego używa się go w małym nadmiarze (np. 1,2 równoważnika), aby wszystko przereagowało w rozsądnym czasie
jest jednocześnie zużywane jako źródło atomów wodoru oraz propagator rodników, dlatego używa się go w małym nadmiarze (np. 1,2 równoważnika), aby wszystko przereagowało w rozsądnym czasieWarto zadać sobie pytanie – skąd bierze się ta selektywność? Częściową odpowiedź przynosi nam termodynamika. Energia wiązania  jest o około
jest o około  większa od energii wiązania
większa od energii wiązania  , a energia wiązania
, a energia wiązania  jest o około
jest o około  większa od energii wiązania
większa od energii wiązania  .
.
Nie jest to jednak to zastosowanie reagenta, które mnie w nim urzekło. Przeprowadźmy prosty eksperyment myślowy: selektywność tej reakcji wynika z dużego zysku energetycznego – rodnikowi nie “opłaca się” reagować z innymi grupami funkcyjnymi. Jednak co jeśli przed oderwaniem wodoru powstały rodnik mógłby ulec jakiejś wewnątrzcząsteczkowej reakcji, w wyniku której powstałby rodnik z niesparowanym elektronem nadal znajdującym się na atomie węgla? Reakcje wewnątrz cząsteczki zachodzą o wiele szybciej niż reakcje pomiędzy cząsteczkami, więc nie ma nic przeciwko temu, jeśli znaleźlibyśmy prostą grupę reagującą w ten sposób.
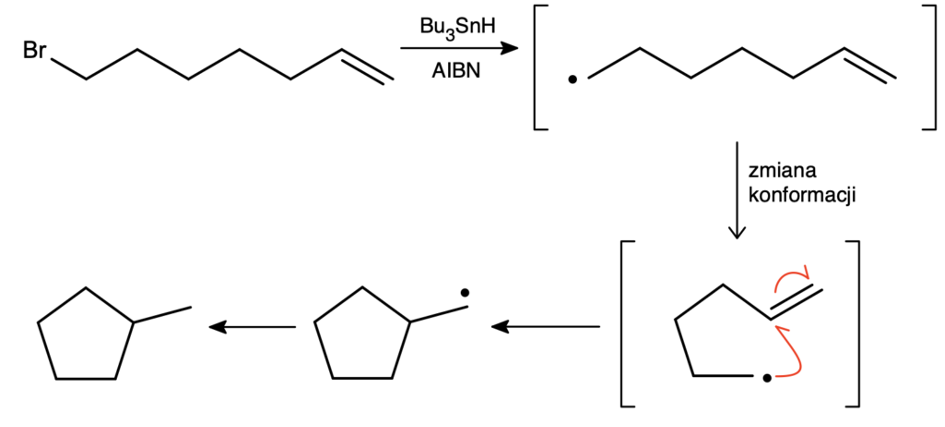
Właśnie tak zachowuje się wiązanie podwójne, które jest bardzo prostą w otrzymaniu grupą! Nasuwa się pytanie: dlaczego nie powstaje stabilniejszy pierścień sześcioczłonowy? Rzeczywiście, jest on nieznacznie stabilniejszy, lecz reakcja ta nie jest odwracalna, a im mniejszy pierścień, tym szybciej się w tej reakcji tworzy; produktu cykloheksanowego powstaje tylko 2%. Aby uzyskać pierścień sześcioczłonowy, należałoby użyć o jeden węgiel dłuższego łańcucha.
Reakcja ta jest potężną i selektywną metodą syntezy pierścieni pięcio- oraz sześcioczłonowych. Zainteresowanych odpowiedzią na pytanie „dlaczego nie powstają mniejsze pierścienie?” zachęcam do zapoznania się z regułami Baldwina.
Pełny potencjał tej reakcji można dostrzec przy syntezie naturalnych związków policyklicznych – motyw sąsiadujących ze sobą pierścieni pięcioczłonowych i sześcioczłonowych jest niezwykle popularny w steroidach. Jednym z takich związków jest trójcykliczny hirsuten z czterema centrami chiralności:
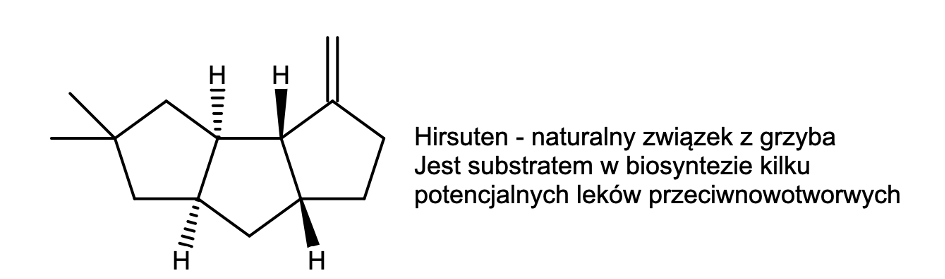
Jedną z pierwszych i zarazem najbardziej eleganckich metod syntezy tego związku opracowano w 1985 roku. Ostatnim i kluczowym etapem była właśnie niezwykle pomysłowa cyklizacja rodnikowa, wykorzystująca szybkie powstawanie pierścieni pięcioczłonowych:
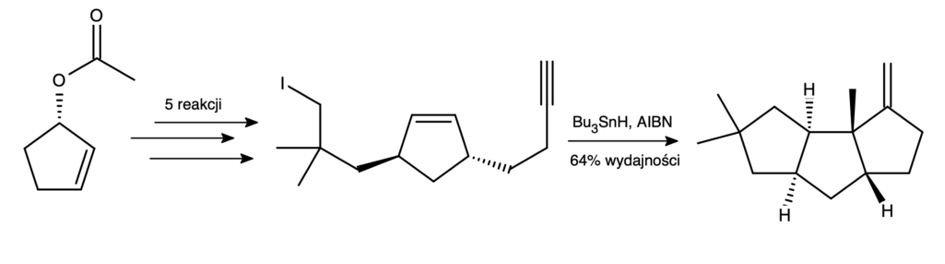
Mechanizm cyklizacji jest analogiczny do poprzedniego:
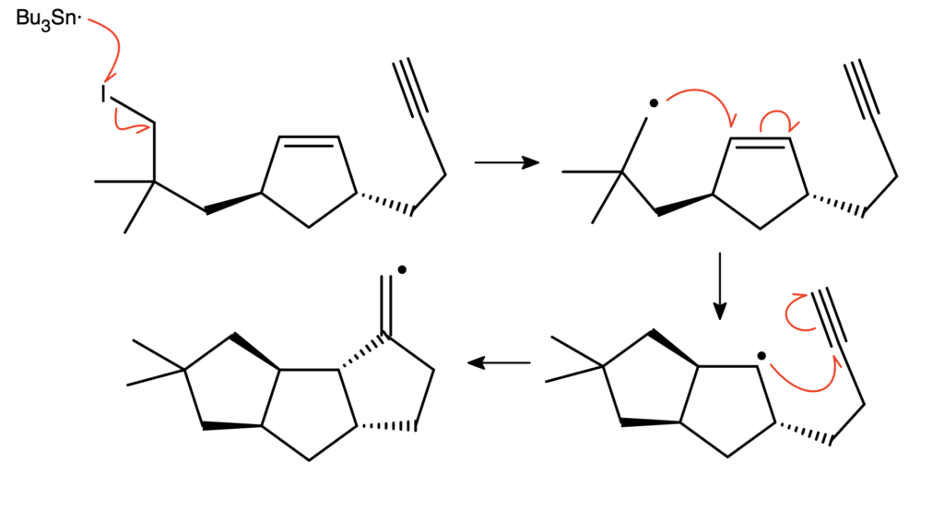
Ostatni rodnik winylowy odrywa wodór od , dając hirsuten
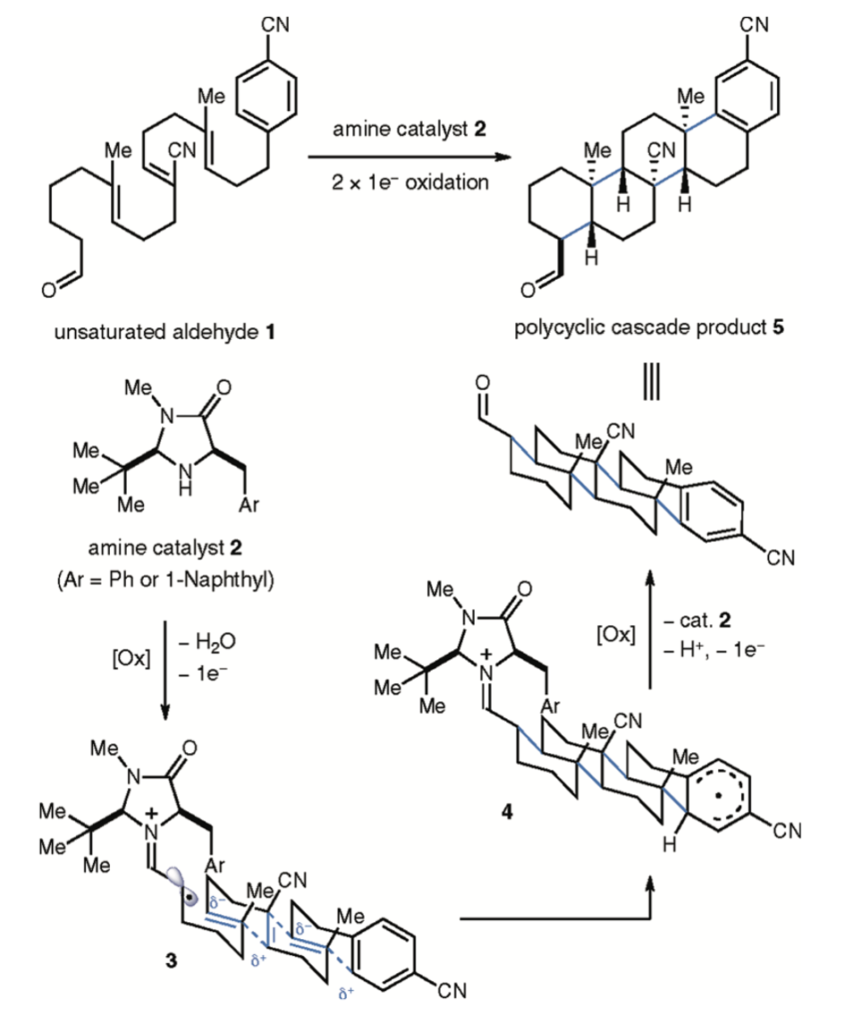
Tego typu kaskadowe cyklizacje otwierają wiele możliwości w syntezie złożonych związków policyklicznych. Jednym z większych ostatnich osiągnięć chemików w tej dziedzinie jest katalizowana aminą cyklizacja, tworząca z achiralnego związku nowy, zawierający 7 centrów stereogenicznych o określonej konfiguracji:
Chemia cynoorganiczna otwiera znacznie więcej możliwości, niż same cyklizacje i redukcje. Od dawna duże zainteresowanie budzą reakcje multikomponentowe (w skrócie MCR) – czyli, jak sama nazwa wskazuje, takie, w których biorą udział więcej niż dwie cząsteczki. Flagowym przykładem jest reakcja Mannicha:
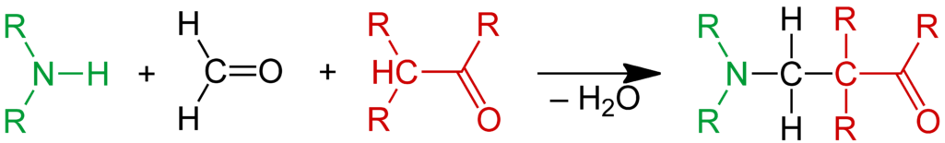
Ta elegancka metoda działa ze względu na idealnie dobraną reaktywność poszczególnych komponentów. Zarówno amina, jak i keton w formie enolowej są nukleofilami, chociaż oczywiście, mogą ze sobą reagować, tak jak dzieje się to podczas aminowania redukcyjnego. Właśnie w takich przypadkach, gdy mamy dobrze znaną reakcję z cząsteczkami o podobnych do siebie właściwościach, możemy “dorzucić” do nich trzeci związek o kompletnie innym charakterze – np. bardzo elektrofilowy formaldehyd. Pełni rolę pewnego pośrednika – aminą reagując z nim tworzy wysoce reaktywny kation iminiowy, który jest chętnie atakowany przez enol.
Podobne rozumowanie zastosowano w przypadku poniższej reakcji rodnikowej:
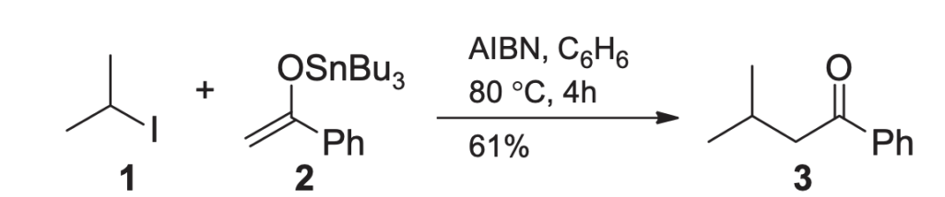
Inicjator rodnikowy odrywa jod i generuje rodnik alkilowy, który następnie atakuje enol cyny. Na koniec dochodzi do eliminacji rodnika tributylocynowego który odtwarza cykl, a przy tym do wytworzenia odpowiedniego ketonu.
Nie jest to jednak najlepsze dobranie – zarówno rodnik alkilowy jak i enol są z natury nukleofilami. Idealnym rozwiązaniem jest dodanie jako pośrednika ubogiego w elektrony alkenu – na przykład akrylonitrylu:
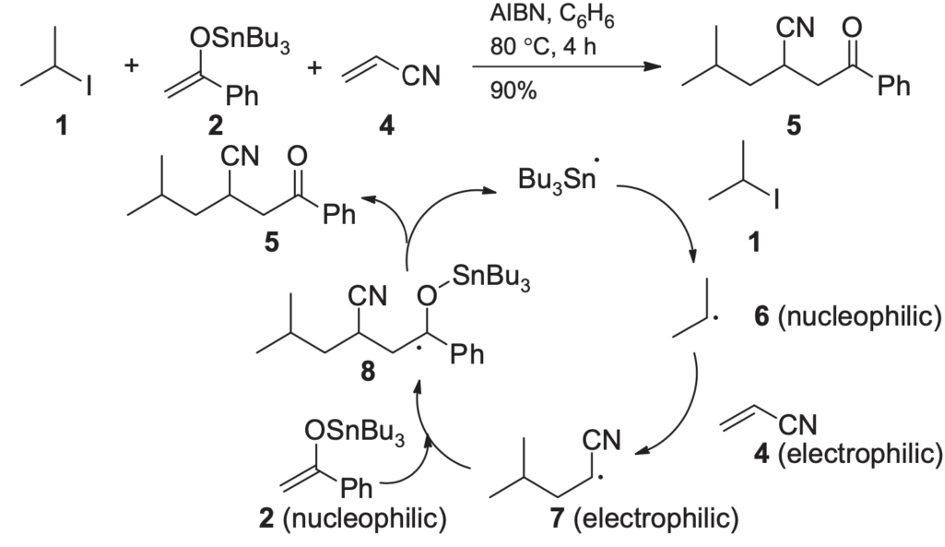
Dzięki starannemu dobraniu reaktywności, możliwe jest nie tylko zwiększenie wydajności reakcji, ale i stopnia złożenia otrzymanego związku – bardziej sfunkcjonalizowane związki są „ciekawsze” syntetycznie.
Selektywność taka wykorzystywana jest też w przemysłowej syntezie wielu polimerów – stosując alken bogaty w elektrony oraz drugi w nie ubogi, otrzymujemy kopolimer naprzemienny, czyli taki, w którym różne mery występują na zmianę, a nie losowo, jak w polimerze statystycznym:
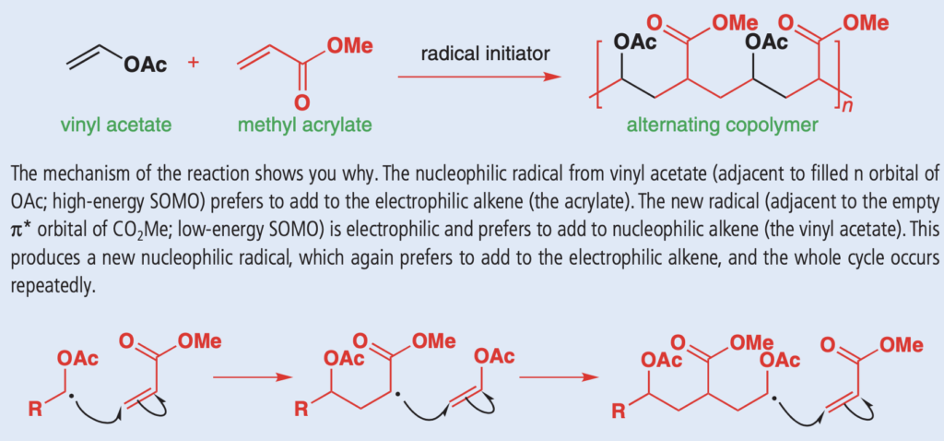
Chemia reakcji rodnikowych jest niezwykle złożona i przedstawienie jej „całości” w policzalnej ilości wpisów jest zwyczajnie niemożliwe – mam jednak nadzieję, że ta krótka seria przedstawiła chociaż jej ogólny zarys. Zachęcam do dalszego, samodzielnego odkrywania reakcji rodnikowych, zaczynając na przykład od tych indukowanych octanem manganu(III) czy jodkiem samaru.